Introduction
The quality assurance of biological research is becoming an increasingly important issue in various research settings. NGS (next-generation sequencing)-based contaminant detections offer promising diagnostics to assess the presence of contaminants. Because biological resources are frequently contaminated by multiple microorganisms, researchers need careful attention to intra- and interspecies sequence similarities.
OpenContami (OCT) investigates the origin of sequenced reads from user-uploaded BAM files and provides highly probable microbial contaminants present in the BAM files that may be contaminated by the laboratory reagents, sequencer carryovers, cross-contamination, etc. The OCT performs the alignment of user-uploaded NGS reads with over 11,300 microbe species genomes; we compiled a database (DB) of microbe genomes based on the RefSeq complete genomes of bacteria, fungi, and viruses and developed an algorithm that exploits unique and multiple hits (Park et al., BMC Biol. 2019). Moreover, the OCT has processed large publicly available NGS data sets, thereby shaping contaminant distribution. This distribution is used as a reference for user-uploaded data and is updated continuously by incorporating the analytical results of open-shared user data and public data sets.
The OCT system utilizes the GUIs (graphical user interfaces) of OpenLooper (OLP), which include email-based communication, account registration, and data manipulation. Users can request to run the OCT pipeline via OpenLooper, and the output is managed by OpenLooper (see Figure 1).
[Figure 1. Schematic overview of the OpenContami (OCT) system]
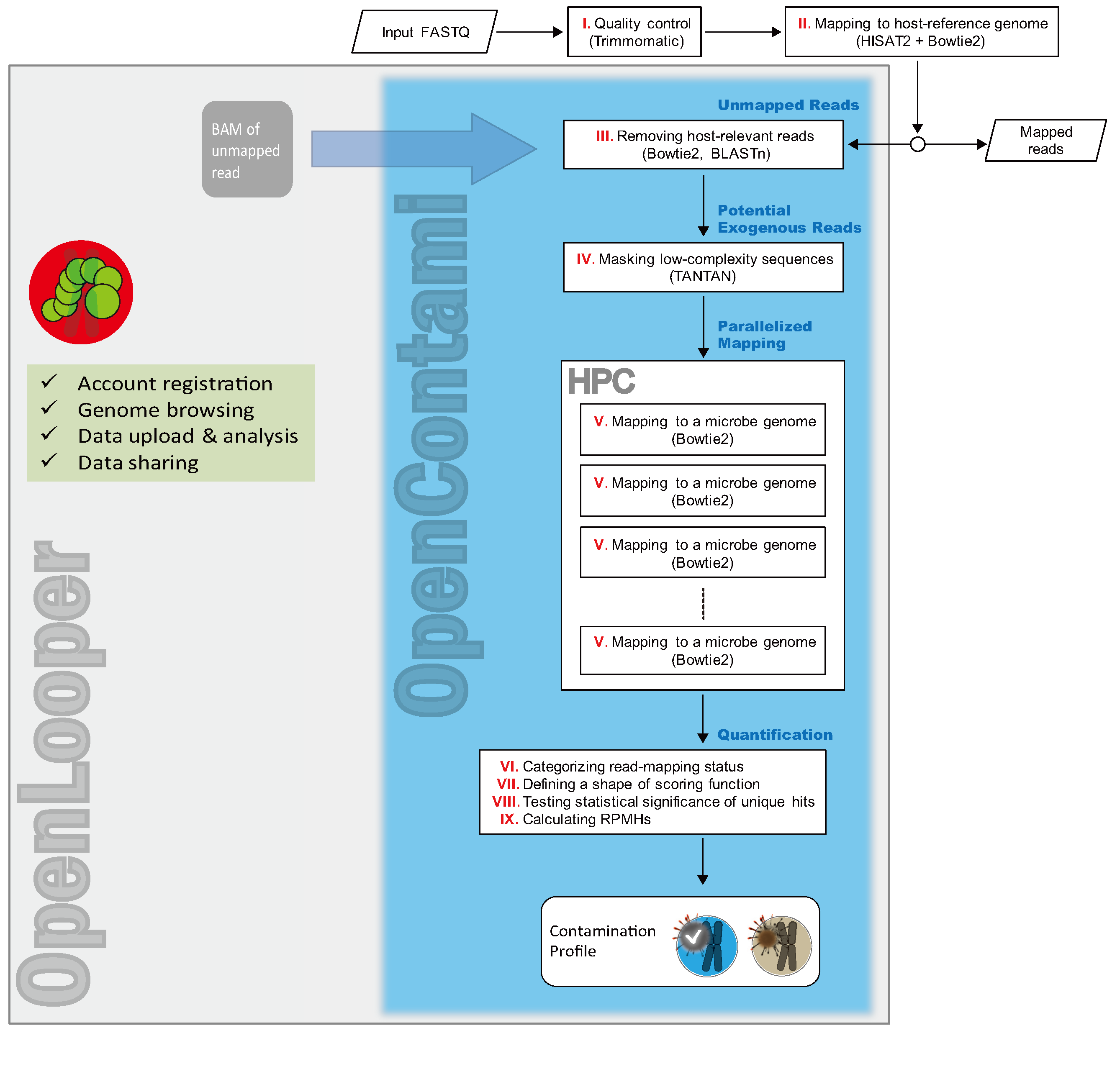
The community-wide effort is indispensable for establishing effective decontamination We hope that the OCT improves our understanding of how and why microbial species infect and contaminate host cells and the impact on the interpretation of experimental results.
Details of the methods can be found in (Park et al., BMC Biol. 2019).
Features
Via OpenContami, the users can do
- Finding and categorizing microbe-originated reads
- Quantifying the microbial NGS reads
- Comparing your data with the OCT records
- Opening your data to the public domain
- Sharing your data with other users
- Browsing the OCT records
- Contributing to the community effort
How to Use (step by step)
- The users have to perform mapping NGS reads to the host reference genome using alignment tools such as Bowtie2, bwa, star, Hisat2.
# run hisat2 for mapping SE (single-end) reads to hisat2-idx %>hisat2 -x hisat2-idx_of_HOST -U input.fastq -S output.sam # or run hisat2 for mapping PE (paired-end) reads to hisat2-idx %>hisat2 -x hisat2-idx_of_HOST -1 R1.fastq -2 R2.fastq -S output.sam # convert SAM to BAM and sort it %>samtools view -bo output.bam output.sam %>samtools sort -@ 3 -o output.sorted.bam output.bam
- The users have to count the host-mapped reads and preparing the BAM that includes host-unmapped reads only.
# for the SE reads: '4'; read unmapped %>samtools view -cF 4 output.sorted.bam # for the PE reads: '132'; read unmapped and second in pair %>samtools view -cF 132 output.sorted.bam # pick up unmapped reads (SE): '4'; read unmapped %>samtools view -bf 4 -o unmapped.output.bam output.sorted.bam # pick up unmapped R1 reads (PE): '69'; read paired and unmapped and first in pair %>samtools view -bf 69 -o unmapped.output.bam output.sorted.bam # sort %>samtools sort -@ 3 -n unmapped.outout.bam -o unmapped.output.sorted.bam # please CONFIRM that 'unmapped.output.sorted.bam' has unmapped reads %>samtools view -cf 4 unmapped.output.sorted.bam
The users need to complete Step 1 and Step 2 in their local environment.
If the input BAM contained zero unmapped reads, the OCT reports a failure message to you.
To know the meaning of SAM flags, please access here
Note that the OCT requests to input the number of host-mapped reads for calculating RPMH (reads per million host-mapped reads). This is because the BAM is recommended to include only unmapped reads and we cannot get the total number of host-mapped reads to be used for calculating RPMH.
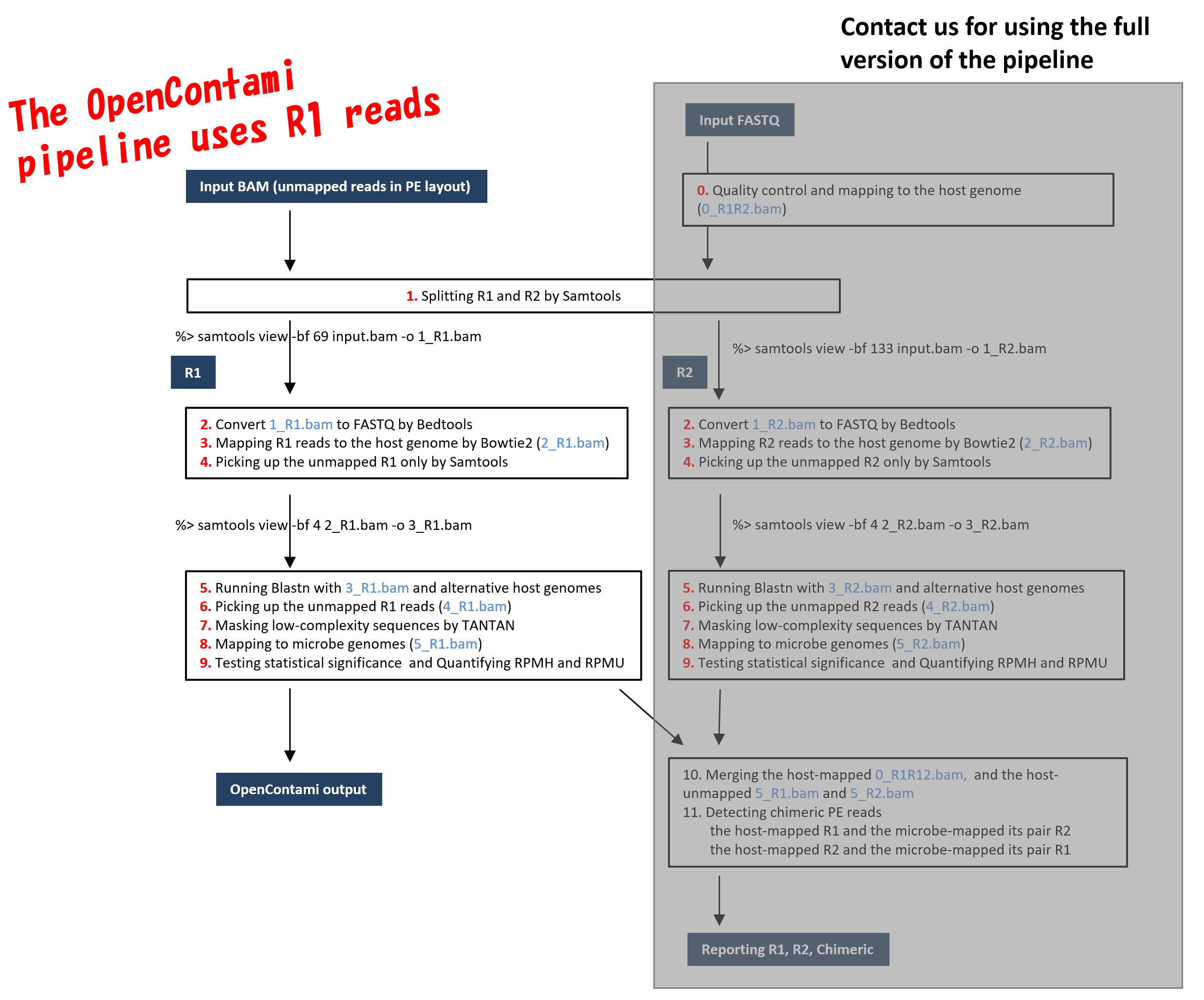
The OCT uses only R1 of PE reads in the current version for reducing computational resources (see Figure 2). If the users want to analyze R1 and R2 simultaneously, contact us.
[Figure 2. Overview of the pipeline dealing with PE (paired-end) NGS reads]
- Register an OpenLooper account
- Login to OpenLooper
- Upload and annotate BAM files (e.g. 'unmapped.output.sorted.bam' above mentioned)
- Select the tab "2.Experiment" of Annotation Edit and set "Reference Genome" to "Others" (other organisms, or unmapped)
- Select the tab "4.Tools" of Annotation Edit and set "OpenContami (OCT) Configuration"
The number of host-mapped reads counted at Step 2 must be written in "3. How many R1 reads were mapped to the host genome". - Submit to the OpenContami pipeline
The OCT contacts the user via email when the submitted job has been finished.
Note that a user can submit a limited number of jobs.How to use OpenContami (OCT) pic.twitter.com/RDKe7sSC0K
— OpenLooper (@w3olp) January 7, 2021
RPMH and RPMU
The microbe-originated reads are normalized in the unit of RPMH (reads per million host-mapped reads) and RPMU (reads per million host-unmapped reads). RPMH represents the number of microbial reads mappable to known microbial genomes when a million host reads have been sequenced, while RPMU represents that when a million origin-unknown reads present. To calculate RPMH, the users need to fill the total number of host-mapped reads (see Step2 in "How to Use").
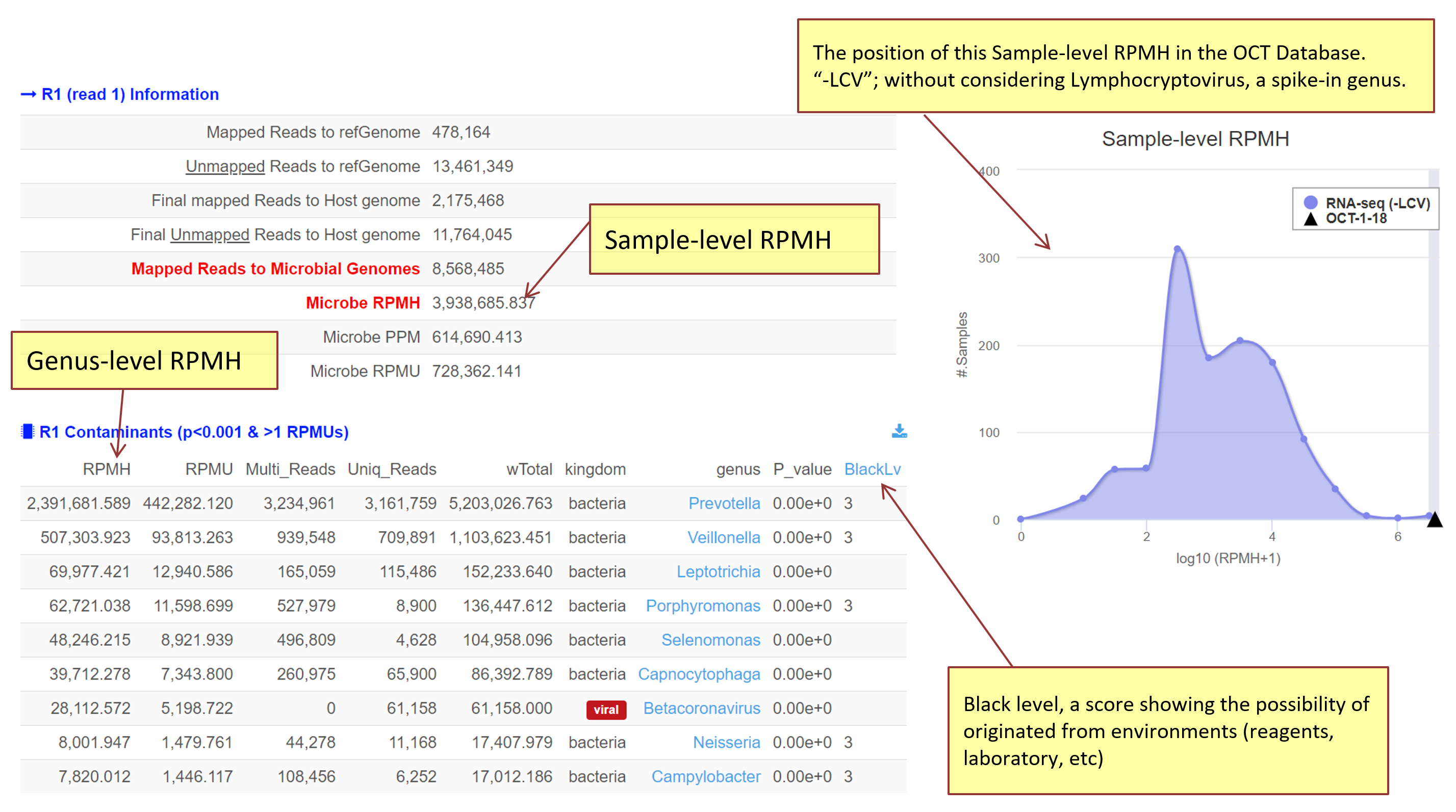
There are two types of RPMH, one for a sample and the other for each contaminant detected (see Figure 3).
Sample-level RPMH: For example, "RNA-seq sample A has 1000 RPMH" means that when 1 million host reads sequenced in the sample A, 1000 reads were mapped to microbial genomes uniquely and/or repeatedly.
Genus-level RPMH: For example, "Bacteria B in RNA-seq A has 1000.465 RPMH” means thatwhen 1 million host reads sequenced in the sample A, 1000.465 weighted reads were found for the bacteria B. The weight is based on the empirical exponential scoring function in (Park et al., BMC Biol. 2019).
Also, the RPMU has sample- and genus-level values. The RPMU does not use the number of host-mapped reads.
BlackLv
Each genus detected by the OCT pipeline is listed along with its BlackLv (black level) score (see Figure 3). The BlackLv is a score for genus inferred by integrative analysis of negative blank controls (Blank-seq): a higher score implies that a genus is more frequently observed in negative controls, suggesting that the target cells are not contaminated or infected by the genus. The current version (v1, 2020/10/20) includes 157 datasets of negative blank controls (PRJEB21503, PRJEB36408, PRJEB7055), Table S8 in (PMC7500457), and Table 1 in (PMC4228153).
- Score 5: OCT detected from at least two PRJEB (=3), and listed at both Tables (1+1)
- Score 4: OCT detected from at least two PRJEB (=3), and listed at either Table (=1)
- Score 3: OCT detected from at least two PRJEB (=3), and listed at neither Table (=0)
- Score 2: OCT did not detect, but listed at both Tables (=2)
- Score 1: OCT did not detect, but listed at either (=1)
- Score 0: otherwise
[Figure 3. Structure of the output web-page]
About Output
For the overview of input file,
- Raw Reads: how many reads in the input
- Number of Raw Reads after QC Filtering: how many reads retained after QC filtering
- (only for PE) Mapped Reads to refGenome (Paired-End): host-mapped reads
- (only for PE) Unmapped Reads to refGenome (Paired-End): host-unmapped reads
For either R1 or R2,
- Mapped Reads to refGenome: how many reads mapped to the host reference genome
- Unmapped Reads to refGenome: how many reads unmapped to the host reference genome
- Final mapped Reads to Host genome: how many reads mapped to the host reference and alternative genome sequences
- Final Unmapped Reads to Host genome: how many host-unmapped reads
- Mapped Reads to Microbial Genomes: total number of reads mapped to microbial genomes
- Microbe RPMH: sample-level RPMH (reads per million host-mapped reads)
- Microbe PPM: sample-level parts per million
- Microbe RPMU: sample-level RPMU (reads per million host-unmapped reads) sample-level parts per million
- (only for PE) Chimeric Reads (Host-Microbe): total number PE reads which one was mapped to the host genome and the other was mapped to microbes
- (only for PE) Chimeric RPMH: chimeric reads in the unit of RPMH
- (only for PE) Chimeric PPM: chimeric reads in the unit of PPM
For each genus,
- RPMH: genus-level reads per million host-mapped reads
- RPMU: genus-level reads per million host-unmapped reads
- Multi_Reads: total number of reads mapped to multiple microbial genera
- Uniq_Reads: total number of reads mapped to a unique microbe genus
- wTotal: the sum of weighted score
- kingdom: bacteria, fungi, viral
- genus: taxonomy
- P_value: the significance of the Uniq_Reads comparing with the ensamble of Uniq_Reads derived from random sampling
- BlackLv: score of black level inferred in negative controls
About Database
The Sample-level RPMH estimated for a user uploaded data is displayed in the distribution of Sample-level RPMHs of the database (DB), indicating how frequently the user’s RPMH is observe (see Figure 3). To build the DB, we analyzed publicly available human NGS read sets such as 1000 Genomes (Genomes Project, et al., 2015), GEUVADIS (Lappalainen, et al., 2013), ENCODE (Consortium, et al., 2020), and CCLE (Ghandi, et al., 2019) and more.
To build the DB, we analyzed publicly available human NGS read sets such as 1000 Genomes, GEUVADIS, ENCODE, CCLE, and more. The DB will be updated by the user's data opened to the public domain.
Note that some of cell-line-derived samples include a higher level of known microbes, such as LCV (lymphocryptovirus; HHV4) and PhiX174microvirus (Illumina spike-in). We excluded LCV in the RPMH distribution as denoted "-LCV".
Download
- Microbe Genome Dataset
- Bacteria: BAC.tar.gz (47Gb)
- Fungi: FUNGI.tar.gz (8.5Gb)
- Viruses: VIR.tar.gz (358Mb)
- Resource used in Park et al., BMC Biol. 2019
- GitHub:
- Reversion Test Source: RNA.tar.gz (2.5Gb)
- List of Kingdom, Genus, RefSeq, Species IDs: MAP_Kingdom_Genus_Species.txt.gz (2.2Mb)
Bug Reporting
We always appreciate bug reports and feedback. When you report a bug, please include the following information;
- Reporter: Your name and email address
- Date and time: When you saw the bug
- URL: The page URL on which the bug occurred
- Screen images: Attaching screen capture images
- Expected and actual results: What our system did contradicting in expectation

How to Cite OCT
OpenContami algorithm
-
Park SJ, Onizuka S, Seki M, Suzuki Y, Iwata T, Nakai K.
A systematic sequencing-based approach for microbial contaminant detection and functional inference
BMC Biol. 2019 Sep 13;17(1):72. doi: 10.1186/s12915-019-0690-0
PMID: 31519179
OpenContami web application
-
Park SJ and Nakai K.
OpenContami: A web-based application for detecting microbial contaminants in next-generation sequencing data
Bioinformatics 2021. doi: 10.1093/bioinformatics/btab101
PMID: 33576798

 Log out (
)
Log out (
)